Manage Your Risk: Monitoring the Environment of Aseptic Processes

An interview with Lisa G. Lawson, conducted by Alina Shrourou, BSc
Please define the meaning of “risk” from the perspective of environmental monitoring.
Risk assessment consists of the identification of hazards and the analysis and evaluation of risks associated with exposure to those hazards. The risk assessment of particle and microbiological samples should consider:
- Highest risk of contamination of the product and its components (study of the airflows and exhaust grid)
- Personnel operations
- Material and personnel flows
- Cleaning and sanitizing of environments and equipment
 Credit: CoolaM/ Shutterstock.com
Credit: CoolaM/ Shutterstock.comWhat are the stages of an aseptic process and which point is most vulnerable to contamination?
Two clean areas are of particular importance to sterile drug product quality: the critical area and the supporting clean areas associated with it.
Critical areas are where the product is most susceptible to contamination. Most commonly, this is during manipulation of sterile materials before and during filling and closing operations (eg, asceptic connections or the addition of sterile ingredients).
Here, a sterilized drug product becomes exposed to the environment and presents the opportunity for contamination – if this occurs, the product will no longer be sterilized when it reaches the sterile container. Therefore, we take extra care in ensuring the critical area is especially designed to ensure product sterility.
Of course, it is important to maintain product sterility by controlling the environment that aseptic processes take place, at a suitable quality. However, it’s not often considered that an aspect of environmental quality is the particle content of the air.
Particles can not only enter and contaminate a product extraneously, but can also contaminate a previously sterile drug product by acting as a vehicle for microbial contamination. Therefore, the environment of which aseptic processes take place should contain suitably designed air handling systems to reduce the particle content within a critical area.
Please outline the FMEA method for microbial monitoring. How does it identify and evaluate risk?
Quality Risk Management (QRM) activities are performed using systematic processes designed to coordinate, facilitate and improve science-based decision-making with respect to risk.
The analysis of the risk assessment is carried out using the FMEA method – the typical mathematical relationship that links the "Risk" RPI (Risk Priority Index) with the "Probability of contamination", the "severity" of the expected damage, and the "Detectability" of the damage, summarized as: RPI = P1 x P2 x S x D.
- P1 = The estimate of the probability of environmental contamination
- P2 = The estimate of the probability of product contamination
- S = Severity which defines the importance of danger on the basis of its consequences
- D = The ability to detect any particles present in the environment through the use of different monitoring methods
How are levels of risk measured and what are the criteria for these?
The following is a typical matrix used to evaluate risk:
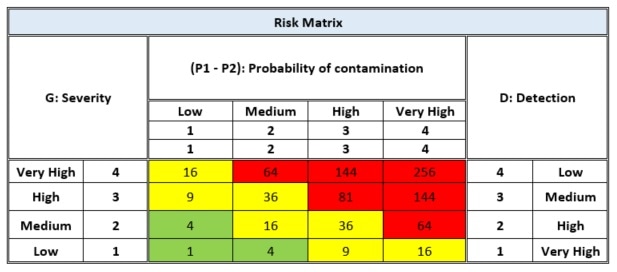
Therefore, the risk for controlled environments is defined as:
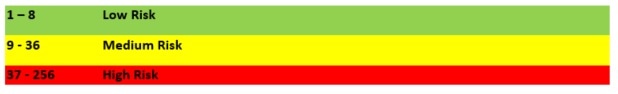
Following this analysis, you proceed to the next step which involves risk control. Risk control is a decision-making activity designed to reduce or accept risks of the existing microbiological/particle control.
The purpose of risk control is to reduce the risk to an acceptable level. Risk reduction includes all the actions to be taken to reduce the probability of contamination value or increase detectability value by employing specific methods of sampling.
What service can Particle Measuring Systems provide for sterility assurance?
Particle Measuring Systems provide our customers with the tools to ensure sterility of their products through comprehensive analysis and robust final documentation. We work closely with pharmaceutical companies to promote sterility education and successful procedure implementation.
We provide support on sterility assurance deviations, pharmaceutical process design evaluation and risk analysis, US and international regulatory audit support, and quality crisis management (warning letter and 483 observation response support).
If a risk is identified, how can the environment be altered to eliminate it? How easy is it to do this?
The purpose of risk control is to reduce the risk to an acceptable level. Risk reduction includes all the actions to be taken to reduce the probability of contamination value or increase detectability value by employing specific methods of sampling. These sampling methods may include: volumetric air sampling, settle plates, surface sampling and total particle air sampling
Do Particle Measuring Systems offer any support to help customers change their production process to eliminate any risks identified by using the service?
We offer support in many areas including but not limited to:
Training and certification in:
- Fundamentals of Microbiology
- Personnel Hygiene GMP practices
- Contamination Control
- Cleanroom qualification training
- Cleaning and Sanitization GMP practices
- Alternative Microbiological Methods training
- Risk Assessment use in contamination control
- Media Fill Requirements training
- FDA and EU guideline interpretation training
- Regulatory Update training
- Any other training topic that can customized to meet customer needs
Sterility Assurance issues handling including:
- Development of a Corporate Sterility Assurance program
- Training, development or qualification for sterilization and/or depyrogenation
- Aseptic Process Management and/or facility design projects which may include:
- Review of production deviations, non-conformities or other investigations
- Validation of microbial sampling systems and isolators for sterility testing
Inspection Support and Management including:
- Resolution of FDA 483, EMA or local authority compliance deviations
- On-site support during regulatory inspections
Quality By Design (QbD)and Process Analytical Technology (PAT) support
- Providing advice on how QbD/PAT may be incorporated in the business system
- Applying comparability protocols and other regulatory strategies for deployment
Process and Methods Evaluation and/or Risk Assessment including:
- Performance of formal risk analysis using Failure Modes and Effects Analysis (FMEA), Hazard Analysis (HACCP) or other Risk Analysis Tool
- Evaluation of laboratories, production and quality operations
- Development of programs or procedures
- Review of data trends and recommendation of alert and action levels
Pharmaceutical Microbiology Support including:
- Environmental Monitoring evaluation and development
- Evaluation and development of protocols and cleaning procedures
- Evaluation and development of procedures, policies and standards for media fill
- Microbiological Data Deviations (MDD) and analysis of Out-of-Specification (OOS)
- Program Development and review of microbiological data deviations and OOS
- Implementation of corrective action plans for reducing and/or eliminating MDD or OOS
Production and Quality Assurance Support including:
- Process Optimization, or project management assistance
- Comparability Protocols for process/analytical changes
- Cleaning and sanitization process analysis including practices, procedures and rationales/rotations
- Development of Quality Management System (QMS)
- Evaluation of systems based inspection compliance programs
- Support implementation of quality problem solving tools
- Assist in investigation resolution, reduction and root cause analysis
Quality Control Support including:
1.Laboratory Management
- Optimization of laboratory policies, procedures, equipment and methods
- Sterility test analysis and evaluation of positive sterility failure tests
2.Development of rapid methods for sterility testing
How often are sterility assurance methods reviewed or improved?
Once risk control measures have been identified, it is important to ensure all stakeholders are aware of the risks identified and are in alignment with the mitigation measures employed to reduce risk. Therefore, the last steps in the QRM process are risk communication and periodic risk review.
Risk communication is the sharing of information about risk and risk management between decision makers and others. Parties can communicate at any stage of the risk management process. The output/result of the quality risk management process should be appropriately communicated and documented.
Appropriate systems should be in place to ensure that the output of the QRM process is periodically monitored and reviewed, as appropriate, to assess new information that may impact the original QRM decision. Examples include changes to control systems, equipment and processes, suppliers or contractors and organizational restructuring.
What does the future hold for the development of environmental risk assessment? Will processes become completely automated?
The future for environmental risk assessment is to automate processes as much as possible to further reduce the risk of personnel contamination, as human interventions pose the greatest particulate and microbiological contamination risk to the product.
Where can readers find more information?
Please find more information on our Advisory services at the following link: https://www.pmeasuring.com/en/service-and-support/consultancy-training
To view our full line of innovative environmental monitoring products, go to: https://www.pmeasuring.com
About Lisa Lawson
Lisa G. Lawson has over 25 years’ experience in supporting large and small pharmaceutical companies in contamination control, including cleaning and disinfection strategies, aseptic manufacturing and use of risk-based approaches to microbiological quality challenges.
Lisa spent 16 years working for Amgen in microbial and mammalian cell culture manufacturing support and helped gain commercial FDA and international regulatory approval of the EPO/NESP manufacturing facility in Longmont, CO.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices and treatments.






















.png)










No hay comentarios:
Publicar un comentario