

No medical product safety issues to report.
Comunicaciones de la FDA sobre la seguridad de los medicamentos en español
Descargo de responsabilidad: La FDA reconoce la necesidad de proporcionar información importante sobre seguridad de los medicamentos en idiomas distintos al inglés. Hacemos lo mejor posible para proporcionar versiones en español precisas y oportunas de nuestras Comunicaciones de Seguridad de Medicamentos. Sin embargo, en caso que existiera discrepancias entre las versiones en inglés y la de español, la información contenida en la versión en inglés es la que se considera como versión oficial. Si tiene alguna pregunta, por favor contáctese con Division of Drug Information en druginfo@fda.hhs.gov. Comunicaciones de la FDA |


FDA knows the major public health consequences that can result from drug shortages. These shortages occur for many reasons including manufacturing and quality problems, delays and discontinuations. When issues are discovered, FDA works closely with the company to address risks involved to prevent harm to patients. FDA also considers the impact a shortage would have on patient care and access and works with the company to restore supplies while also ensuring safety for patients. More information
|
Drug Shortages Voluntarily Reported by Manufacturers During the Past 2 Weeks:
Drug Discontinuation Voluntarily Reported by Manufacturers During the Past 2 Weeks:
Information about blood and biologic shortages, resolved shortages, and discontinuations
La FDA reconoce las consecuencias significativas para la salud pública que pueden resultar de la escasez de medicamentos y hace un gran esfuerzo dentro de sus facultades legales para abordar yprevenir la escasez de medicamentos. La escasez se produce por muchas razones, incluyendo problemas de fabricación y calidad, retrasos y discontinuación del producto. Cuando los problemas son descubiertos por la empresa o el público y reportados a la FDA o se descubren por inspecciones de la FDA, la FDA trabaja en estrecha colaboración con la empresa para hacer frente a los riesgosinvolucrados y evitar daños a los pacientes. La FDA también considera el impacto que una escaseztendría en la atención médica del paciente y al acceso del producto y trabaja con la empresa pararestablecer el suministro al tiempo que garantiza la seguridad de los pacientes. Más información
|

FDA allows marketing of test to aid in the detection of certain leukemias and lymphomas
FDA has allowed marketing of ClearLLab Reagents (T1, T2, B1, B2, M), the first agency authorized test for use with flow cytometry to aid in the detection of several leukemias and lymphomas, including chronic leukemia, acute leukemia, non-Hodgkin lymphoma, myeloma, myelodysplastic syndrome (MDS) and myeloproliferative neoplasms (MPN). “This represents a major step forward for the hematology-oncology community,” said Alberto Gutierrez, Ph.D., Director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health. “Laboratories and health care professionals now have access to an FDA-validated test that provides consistent results to aid in the diagnoses of these serious cancers.” More information
FDA approves rituximab plus hyaluronidase combination for Treatment of FL, DLBCL and CLL
FDA has granted regular approval to the combination of rituximab and hyaluronidase human (RITUXAN HYCELA, Genentech Inc.) for adult patients with follicular lymphoma, diffuse large B-cell lymphoma, and chronic lymphocytic leukemia. The approval provides patients a subcutaneous route of rituximab administration that shortens the administration time to 5 to 7 minutes as compared to intravenous infusion that can take several hours. This new product also provides for flat dosing. More information
FDA grants regular approval to dabrafenib and trametinib combination for metastatic NSCLC with BRAF V600E mutation
FDA has granted regular approvals to dabrafenib and trametinib (TAFINLAR® and MEKINIST®, Novartis Pharmaceuticals Inc.) administered in combination for patients with metastatic non-small cell lung cancer (NSCLC) with BRAF V600E mutation as detected by an FDA-approved test. These are the first FDA approvals specifically for treatment of patients with BRAF V600E mutation-positive metastatic NSCLC. More information |
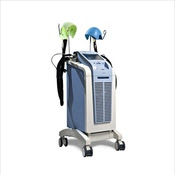
FDA clears expanded use of cooling cap to reduce hair loss during chemotherapy
FDA has cleared the expanded use of a cooling cap, DigniCap Cooling System, to reduce hair loss (alopecia) during chemotherapy. This is the first cooling cap cleared by the agency for use in cancer patients with solid tumors. Hair loss is a common side effect of certain types of chemotherapy and is commonly associated with the treatment of most solid tumor cancer. The DigniCap Cooling System is indicated to reduce the frequency and severity of hair loss during chemotherapy in solid tumor cancer patients in which alopecia-inducing chemotherapeutic agents and doses are used. It is a computer-controlled system used during treatment. More information
FDA approves first subcutaneous C1 Esterase Inhibitor to treatFDA has approved Haegarda, the first C1 Esterase Inhibitor (Human) for subcutaneous (under the skin) administration to prevent Hereditary Angioedema (HAE) attacks in adolescent and adult patients. The subcutaneous route of administration allows for easier at-home self-injection by the patient or caregiver, once proper training is received. HAE, which is caused by having insufficient amounts of a plasma protein called C1-esterase inhibitor (or C1-INH), affects approximately 6,000 to 10,000 people in the U.S. People with HAE can develop rapid swelling of the hands, feet, limbs, face, intestinal tract or airway. These attacks of swelling can occur spontaneously, or can be triggered by stress, surgery or infection. More information
FDA approved betrixaban (BEVYXXA, Portola) for the prophylaxis of venous thromboembolism (VTE) in adult patientsFDA has approved betrixaban (BEVYXXA, Portola) for the prophylaxis of venous thromboembolism (VTE) in adult patients hospitalized for an acute medical illness who are at risk for thromboembolic complications due to moderate or severe restricted mobility and other risk factors for VTE. Approval was based on data from APEX (NCT01583218), a randomized, double-blind, multinational clinical trial comparing extended duration betrixaban (35 to 42 days) to short duration of enoxaparin (6 to 14 days) in the prevention of VTE in an acutely medically ill hospitalized population with risk factors for VTE. The trial randomized 7,513 patients to either betrixaban or enoxaparin treatment. Patients on the betrixaban arm took an initial dose of 160 mg orally on day 1, then took 80 mg once daily for 35 to 42 days and received a placebo injection once daily for 6 to 14 days. Patients on the enoxaparin arm received 40 mg subcutaneously once daily for 6 to 14 days and took a placebo pill orally once daily for 35 to 42 days. More information
|
For information on drug approvals or to view prescribing information and patient information, please visit Drugs@FDA or DailyMed.

View FDA's Comments on Current Draft Guidance page, for a list of current draft guidances and other topics of interest for patients and caregivers.
|

FDA Working to Lift Barriers to Generic Drug Competition, by Scott Gottlieb, M.D., Commissioner of FDA.
Too many patients are being priced out of the medicines they need. While FDA doesn’t have a direct role in drug pricing, we can take steps to help address this problem by facilitating increased competition in the market for prescription drugs through the approval of lower-cost, generic medicines. Over the last decade alone, competition from safe and effective generic drugs has saved the health care system about $1.67 trillion. When generics are dispensed at the pharmacy, the immediate savings to each of us are clear. We could see even greater cost savings if we helped more safe and effective generic drugs get to market sooner, after patent and statutory exclusivity periods have lapsed, by addressing some of the scientific and regulatory obstacles to generic competition across the full range of FDA-approved drugs. These barriers may delay and, in some cases, ultimately deny patient access to more affordable drugs. That’s why we’re working on a Drug Competition Action Plan. As part of this effort, today, we’re announcing in the Federal Register our intent to hold a public meeting on July 18, 2017, to solicit input on places where FDA’s rules – including the standards and procedures related to generic drug approvals – are being used in ways that may create obstacles to generic access, instead of ensuring the vigorous competition Congress intended.
To read the rest of this post, see FDA Voice Blog, June 21, 2017.
|

Statement from FDA Commissioner Scott Gottlieb, M.D., on the importance of the Drug Quality and Security Act and overseeing the safety of compounded drugsIn late 2012, the United States faced the most serious outbreak associated with contaminated compounded drugs in recent history, involving hundreds of people, in many states, who developed fungal infections related to a compounded product. It was an incident that resulted in dozens of deaths.
The fungal meningitis outbreak underscores the need for robust oversight over human drug compounding, the importance of dispensing prescription drugs pursuant to valid prescriptions and the need for strong coordination with state regulatory partners to protect public health.
Since that outbreak and the subsequent enactment of the Drug Quality and Security Act (DQSA) on Nov. 27, 2013, the FDA has devoted significant resources to oversee compounding and implement the compounding provisions of the law. As of June 1, 2017, the FDA has conducted more than 400 inspections, including 109 inspections of outsourcing facilities; issued more than 150 warning letters advising compounders of significant violations of federal law; issued more than 50 letters referring inspectional findings to state regulatory agencies; overseen over 125 recalls involving compounded drugs; and worked with the Department of Justice on a number of civil and criminal enforcement actions.
As part of the implementation of DQSA, we have also issued 21 draft guidances, ten final guidances, three proposed rules, a final rule, and a draft memorandum of understanding. We have taken a risk-based approach to all of these efforts, in order to make sure that we are maximizing the public health purpose of these new provisions relative to the resources we use to achieve them, and any obligations that these new requirements place on market participants. These foundational regulations and guidance documents provide predictability and transparency to compounders, providers, and other enterprises; and inform them of how to comply with the law’s provisions in the most efficient manner.
These efforts are part of our commitment to doing all we can to protect the public from poorly compounded drugs. We will continue to actively oversee drug compounders and, when appropriate, initiate regulatory action as it fulfills the FDA public health mission on behalf of patients.
The FDA, an agency within the U.S. Department of Health and Human Services, promotes and protects the public health by, among other things, assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products for human use, and medical devices. The agency also is responsible for the safety and security of our nation’s food supply, cosmetics, dietary supplements, products that give off electronic radiation, and for regulating tobacco products.
|
FDA unveils plan to eliminate orphan designation backlogFDA has recently unveiled a strategic plan to eliminate the agency’s existing orphan designation request backlog and ensure continued timely response to all new requests for designation with firm deadlines. The agency’s Orphan Drug Modernization Plan comes a week after FDA Commissioner Scott Gottlieb committed to eliminating the backlog within 90 days and responding to all new requests for designation within 90 days of receipt during his testimony before a Senate subcommittee. As authorized under the Orphan Drug Act, the Orphan Drug Designation Program provides orphan status to drugs and biologics that are defined as those intended for the safe and effective treatment, diagnosis or prevention of rare diseases, which are generally defined as diseases that affect fewer than 200,000 people in the United States. Orphan designation qualifies the sponsor of the drug for various development incentives, including tax credits for clinical trial costs, relief from prescription drug user fee if the indication is for a rare disease or condition, and eligibility for seven years of marketing exclusivity upon approval. A request for orphan designation is one step that can be taken in the drug development process and is different than the filing of a marketing application with the FDA. More information
FDA Tackles Drug Competition to Improve Patient AccessFDA is taking two new, important steps to increase competition in the market for prescription drugs and facilitate entry of lower-cost alternatives. The agency published a list of off-patent, off-exclusivity branded drugs without approved generics, and also implemented, for the first time, a new policy to expedite the review of generic drug applications where competition is limited. These actions are among the first taken under the agency’s Drug Competition Action Plan, announced by FDA Commissioner Scott Gottlieb in late May. “No patient should be priced out of the medicines they need, and as an agency dedicated to promoting public health, we must do our part to help patients get access to the treatments they require,” said FDA Commissioner Scott Gottlieb, M.D. “Getting safe and effective generic products to market in an efficient way, being risk-based in our own work and making sure our rules aren’t used to create obstacles to new competition can all help make sure that patients have access to more lower-cost options.” More information

In this section you will find a comprehensive list of all the meetings that the FDA is involved with. The meetings may include advisory committee meetings, public workshops and public conferences that are seeking to hear from patients and caregivers.
Most FDA meetings are free to the public and do not require the public to register. Interested persons may present data, information, or views, orally at the meeting, or in writing, on issues pending before the committee. Other types of meetings listed may require prior registration and fees.
|
Public Meeting: The Hatch-Waxman Amendments: Ensuring a Balance Between Innovation and Access
Date: Tuesday, July 18, 2017
Time: 9 a.m. to 5 p.m.
The purpose of this meeting is to provide the public an opportunity to submit comments concerning administration of the Hatch-Waxman Amendments to the Federal Food, Drug, and Cosmetic Act to help ensure the intended balance between encouraging innovation in drug development and accelerating the availability to the public of lower cost alternatives to innovator drugs is maintained. To provide public comments, please click on https://www.regulations.gov/ and enter the document number: FDA-2017-N-3615. The deadline for submitting comments regarding this meeting is September 18, 2017. More information
Date: Tuesday, July 18, 2017
Time: 9 a.m. to 5 p.m.
The purpose of this meeting is to provide the public an opportunity to submit comments concerning administration of the Hatch-Waxman Amendments to the Federal Food, Drug, and Cosmetic Act to help ensure the intended balance between encouraging innovation in drug development and accelerating the availability to the public of lower cost alternatives to innovator drugs is maintained. To provide public comments, please click on https://www.regulations.gov/ and enter the document number: FDA-2017-N-3615. The deadline for submitting comments regarding this meeting is September 18, 2017. More information
Other meetings:
- Calendar of FDA Sponsored Public Meetings - July 2017
- Calendar of FDA Sponsored Public Meetings - August 2017
- Calendar of FDA Sponsored Public Meetings - September 2017
View FDA's Patient Network Calendar of Public Meetings page for a complete list of meetings and workshops.
For additional information on other agency meetings please visit Meetings, Conferences, & Workshops.
|

Tattoo Removal: Options and Results
That tattoo on your arm of a former flame—the one that seemed like a great idea years ago—is kind of embarrassing today. And your spouse is not too crazy about it either. While state and local authorities oversee the practice of tattooing, inks and pigments used in tattoos are subject to FDA oversight as cosmetics. FDA takes action to protect consumers when safety issues arise related to the inks. At the other end of the tattoo process, FDA also regulates laser devices used to remove tattoos. In recent years, FDA has cleared for marketing several types of lasers for tattoo lightening or removal. More information |
More Consumer Updates
For previously published Consumer Update articles that are timely and easy-to-read and cover all FDA activities and regulated products. More information
For previously published Consumer Update articles that are timely and easy-to-read and cover all FDA activities and regulated products. More information
En Español
La información en esta página es para el público en general, y para profesionales y educadores de salud. Esta información puede ser distribuida y publicada sin previa autorización. En Español
La información en esta página es para el público en general, y para profesionales y educadores de salud. Esta información puede ser distribuida y publicada sin previa autorización. En Español

Barbecue Basics: Tips to Prevent Foodborne IllnessIt’s the season for picnics, cookouts, and other outdoor parties. But eating outdoors in warm weather presents a food safety challenge. Bacteria in food multiply faster at temperatures between 40°F and 140°F, so summer heat makes the basics of food safety especially important. “Fortunately, there are a lot of steps consumers can take to keep family and friends from becoming ill,” says Marjorie Davidson, Ph.D., education team leader in FDA’s Center for Food Safety and Applied Nutrition.
Wash hands. It seems basic, but not everyone does it. Wash hands well and often, with soap and water for at least 20 seconds, especially after using the bathroom and before cooking or eating. if you're in an outdoor setting with not bathroom. use a water jug, some soap, and paper towels. Consider carrying disposable towelettes for cleaning your hands. More information
|
The Safety Reporting Portal
The Safety Reporting Portal (SRP) streamlines the process of reporting product issues to the Food and Drug Administration and the National Institutes of Health. Whatever your role, (manufacturer, health care professional, researcher, public health official, or concerned citizen), when you submit a safety report through this Portal, you make a vital contribution to the safety of America's food supply, medicines, and other products that touch us all. More information
The Safety Reporting Portal (SRP) streamlines the process of reporting product issues to the Food and Drug Administration and the National Institutes of Health. Whatever your role, (manufacturer, health care professional, researcher, public health official, or concerned citizen), when you submit a safety report through this Portal, you make a vital contribution to the safety of America's food supply, medicines, and other products that touch us all. More information
Center for Food Safety and Applied Nutrition
The Center for Food Safety and Applied Nutrition, known as CFSAN, carries out the mission of FDA. The Center provides services to consumers, domestic and foreign industry and other outside groups regarding field programs; agency administrative tasks; scientific analysis and support; and policy, planning and handling of critical issues related to food and cosmetics. More information
The Center for Food Safety and Applied Nutrition, known as CFSAN, carries out the mission of FDA. The Center provides services to consumers, domestic and foreign industry and other outside groups regarding field programs; agency administrative tasks; scientific analysis and support; and policy, planning and handling of critical issues related to food and cosmetics. More information
Food Facts for You
The Center for Food Safety and Applied Nutrition, known as CFSAN, issues food facts for consumers to keep you and your family safe. More information
The Center for Food Safety and Applied Nutrition, known as CFSAN, issues food facts for consumers to keep you and your family safe. More information

Animal Health LiteracyAnimal Health Literacy means timely information for the benefit of all animals and their humans. With continuous communication and outreach, the Center for Veterinary Medicine (CVM) strives to enhance the public trust, promote safe and effective use of the animal health products we regulate, and share our scientific endeavors. CVM provides reliable, science-based information to promote animal and human health. More information and Publicaciones en Español del FDA
Animal and Veterinary UpdatesAnimal and veterinary updates provide information to keep your pets healthy and safe. More information
|

How to Report a Pet Food Complaint
You can report complaints about a pet food product electronically through the Safety Reporting Portal or you can call your state’s FDA Consumer Complaint Coordinators.Please provide as much information as possible in your complaint, such as exact name of product, type of container, lot number, UPC codes, how the food was stored, and purchase date and exact location where purchased. If possible, please save the original packaging until the pet food has been consumed. The packaging contains IMPORTANT information often needed to identify the variety of pet food, the manufacturing plant, and the production date. More information |
FDA’s Center for Tobacco Products celebrates 8th anniversary of tobacco oversightIt has been eight years since the landmark Family Smoking Prevention and Tobacco Control Act was signed into law in June 2009. During this period, the agency has taken many strides towards public health protection, as shown by the CDC’s and FDA’s recent findings from the 2016 National Youth Tobacco Survey. Despite these and other encouraging indications of progress, a great deal of work remains to be done in further reducing the death and disease toll of tobacco. Learn about the 2016 National Youth Tobacco Survey findings.
|

Tips to help avoid "vape" battery explosions
You may have heard that e-cigarettes, or "vapes," can explode and seriously injure people. To help reduce the risk of vape battery overheating, fires, and explosions, the FDA has developed an infographic with five key tips, a video on how to report adverse events to the FDA’s Safety Reporting Portal, and shareable and downloadable content to help spread the word about vape battery safety. |

Comments invited as FDA reviews “modified risk” tobacco product applications
The public can now comment on modified risk tobacco product (MRTP) applications submitted by Philip Morris Products S.A., for its IQOS system and three types of Marlboro Heatstick products. By law, tobacco products may not be marketed as being less harmful or presenting lower risk of tobacco-related disease without a written order from FDA, and there are no authorized MRTPs currently on the market in the United States. You can view related Philip Morris Products S.A. documents as they become publicly available, and comment through Dec. 12. |
Center for Tobacco Products updated safety reporting portalCTP recently updated its Safety Reporting Portal, an online tool for reporting suspected problems with tobacco products. Whether you’re a consumer, manufacturer, clinical investigator, or health professional, let FDA know about products that seem to be damaged, defective, contaminated, or that smell or taste wrong. More information and to report a safety concern online.
How cigarettes are made and how you can make a plan to quitEver wonder why it’s so hard to kick smoking to the curb? Well, we’ve got an answer! CTP created an infographic that explores how cigarettes are made, highlighting how their very design may be an obstacle to quitting. As people across the country pledge to quit smoking, consider sharing this infographic with someone who could benefit.
 |
Missed the last issue of CTPConnect?
The Center also publishes a regular periodical known as CTPConnect, a plainspoken digest with the latest stories from the Center. Sign up today to receive the next issue of CTPConnect and other important updates from CTP directly to your inbox. |

Bad reaction to a cosmetic? FDA needs to know
What do you do if you have a reaction after using a cosmetic product? First, stop using the product and contact your healthcare provider. Next, please report it to FDA.
Here’s why that next step is so important: Cosmetic products aren’t required by law to have FDA approval before they go on the market. Companies that market cosmetics have a legal responsibility to ensure product safety, but FDA can only take action if the product is shown to be unsafe after the product is on the market.
And, because the law doesn’t require cosmetic companies to share customer complaints or other safety information with FDA, voluntary reports from consumers and healthcare providers are one of the best ways for FDA to learn about any problems.
Cosmetics include a range of products people use every day, such as moisturizers, makeup, shampoos and conditioners, face and body washes, deodorants, nail care products, hair dyes and relaxers, and tattoos. More information
|
Recalls and Alerts
To see safety alerts and recent recalls related to cosmetics and other products regulated by FDA. More information
To see safety alerts and recent recalls related to cosmetics and other products regulated by FDA. More information
What to watch for, how to report
You can report an allergic reaction, a rash, redness, burn, hair loss, headache, infection, illness, or any other unexpected reaction, whether or not it required medical treatment. You can also report a bad smell, color change, or other sign of contamination. You can choose the way you’d prefer to report:
You can report an allergic reaction, a rash, redness, burn, hair loss, headache, infection, illness, or any other unexpected reaction, whether or not it required medical treatment. You can also report a bad smell, color change, or other sign of contamination. You can choose the way you’d prefer to report:
- Call an FDA Consumer Complaint Coordinator if you want to speak directly to a person about your problem.
- Complete an electronic Voluntary MedWatch form online.
- Complete a paper Voluntary MedWatch form that you can mail to FDA.

Information about Expanded Access
Expanded access, sometimes called "compassionate use," is the use outside of a clinical trial of an investigational medical product (i.e., one that has not been approved by FDA). FDA is committed to increasing awareness of and knowledge about its expanded access programs and the procedures for obtaining access to human investigational drugs (including biologics) and medical devices. More information |
Learn about what your physician should do before submitting a request for individual patient expanded access use of an investigational medical product, who may be eligible for expanded access, associated costs, FDA contacts and more. Information for Patients
|
Learn about your responsibilities under the expanded access pathway, how to submit a request for expanded access for an individual patient (including for emergency use), which forms to use, FDA contacts and more. Information for Physicians
|




















.png)












No hay comentarios:
Publicar un comentario