
Volume 24, Number 4—April 2018
Dispatch
Direct Whole-Genome Sequencing of Cutaneous Strains of Haemophilus ducreyi
On This Page
Michael Marks1 , Maria Fookes1, Josef Wagner, Rosanna Ghinai, Oliver Sokana, Yaw-Adu Sarkodie, Anthony W. Solomon, David C.W. Mabey, and Nicholas R. Thomson
, Maria Fookes1, Josef Wagner, Rosanna Ghinai, Oliver Sokana, Yaw-Adu Sarkodie, Anthony W. Solomon, David C.W. Mabey, and Nicholas R. Thomson
Abstract
Haemophilus ducreyi, which causes chancroid, has emerged as a cause of pediatric skin disease. Isolation of H. ducreyi in low-income settings is challenging, limiting phylogenetic investigation. Next-generation sequencing demonstrates that cutaneous strains arise from class I and II H. ducreyiclades and that class II may represent a distinct subspecies.
Since 2000, the global prevalence of chancroid, caused by Haemophilus ducreyi, has declined (1). H. ducreyi is an emerging cause of cutaneous ulcers in tropical countries (1–4). Cutaneous lesions of H. ducreyi are difficult to distinguish from other common causes of ulcerative skin disease, such as yaws (3,4), which presents problems in diagnosing yaws and has resulted in the World Health Organization recommending molecular testing of yaws-like lesions (5).
Culturing H. ducreyi is challenging. PCR is usually used for diagnosis (6). Culture requirements limit sequencing and phylogenetic analyses. Traditional phylogenies divide genital strains of H. ducreyi into class I and II clades. Most studies suggest that cutaneous strains of H. ducreyi have diversified from within the class I clade (7,8), and a recent study reported cutaneous strains that appear to arise from class II strains (9). These studies have been limited by the number and geographic spread of samples included.
Next-generation sequencing enables whole-genome sequencing from clinical samples without prior culture, bypassing the culture requirements of H. ducreyi and enabling more detailed phylogenetic analysis. We performed next-generation sequencing on samples collected in previous surveys conducted in the Solomon Islands (in 2013) and Ghana (in 2014) (2,4). In both surveys, skin swab specimens had been collected from persons with chronic ulcerative lesions believed, at the time, to be clinically consistent with yaws. DNA was prepared for the current study from samples with residual material from those original surveys. The London School of Hygiene & Tropical Medicine, Solomon Islands National Health Research, and Kwame Nkrumah University of Science and Technology ethics committees approved these studies.
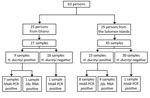
We tested 72 samples from 63 persons (Figure 1). Twenty-five persons (27 samples) had been recruited in Ghana and 38 persons (45 samples) in the Solomon Islands. Median age of participants in the original studies was 9 years (interquartile range 7–11 years); 36 (57.1%) were male. In the original studies, 24 samples had tested positive for H. ducreyi using a 16S rRNA-targeted PCR (2,4): 15 from the Solomon Islands and 9 from Ghana.
In Ghana, samples were collected directly onto dry Dacron swabs. In the Solomon Islands, swab exudate was placed into transport medium (AssayAssure; Sierra Molecular, Incline Village, NV, USA) or onto an FTA Elute Card (Thermo-Fisher Scientific, Waltham, MA, USA). Samples were frozen at −20°C and shipped to the Centers for Disease Control and Prevention (Atlanta, GA, USA) on dry ice for the original laboratory analyses, which included real-time PCR for Trepomema pallidum subspecies pertenue (7) and a real-time 16S rRNA-targeted PCR for H. ducreyi (2,4). After testing, samples were shipped on dry ice to the London School of Hygiene & Tropical Medicine (London, UK) and frozen at −20°C before analysis.
We extracted DNA from residual sample material using QIAamp Mini kits (QIAGEN, Hilden, Germany) (Technical Appendix 1[PDF - 641 KB - 4 pages]). We screened DNA using a quantitative PCR (qPCR) targeting the hhdA gene and 16S rRNA gene sequencing for H. ducreyi (6,10). From samples that tested positive, we selected those with genomic DNA concentration >10 copies/μL for direct (non–culture-based) sequencing.
Genomic DNA was fragmented to an average size of 150 bp and subjected to DNA library creation using established Illumina paired-end protocols (11). We amplified adaptor-ligated libraries and indexed them by PCR. We used a portion of each library to create an equimolar pool and hybridized each pool to custom-made SureSelect RNA baits (Agilent Technologies, Santa Clara, CA, USA; baits based on published sequences of H. ducreyi [12]) (Technical Appendix 1[PDF - 641 KB - 4 pages]). Targets were captured and amplified in accordance with manufacturer’s recommendations. We subjected enriched libraries to standard 75-bp end sequencing (HiSeq 2000; Illumina, San Diego, CA, USA). Samples’ public accession numbers are listed in Technical Appendix 2[PDF - 641 KB - 4 pages] Table 1). We used whole-genome sequence data to estimate phylogenies for H. ducreyi (Technical Appendix 1[PDF - 641 KB - 4 pages]), including publicly available H. ducreyi genomes alongside those obtained in this study.
We identified H. ducreyi in 17 samples by hhdA-targeted qPCR and in 5 additional samples using an assay targeting the rRNA gene. From these 22 positive samples, we obtained 21 (95.5%) complete genomes from 13 persons from the Solomon Islands and 8 from Ghana. Mean coverage of H. ducreyi genomes was 91% (Technical Appendix 2[PDF - 641 KB - 4 pages] Table 1). We found no evidence of sequence heterozygosity that would indicate any participant was infected with multiple distinct strains of H. ducreyi.

Figure 2. Phylogenetic tree of Haemophilus ducreyi genome sequences inferred from mapping using the H. ducreyi 35000HP strain as reference and after removing high-density single-nucleotide polymorphisms regions with Gubbins (3). Included are published...
H. ducreyi sequences fell into both previously defined H. ducreyi clades: class I and class II. To estimate genetic distance between strains, we determined the number of single-nucleotide polymorphisms (SNPs) in pairwise whole-genome comparisons. The average distance between class I and class II sequences was 21,238 SNPs, compared with a maximum pairwise distance of 641 SNPs between class I sequences. We detected 4 major recombination blocks within class I genomes. These regions included the dsrA, tad, and flp loci, associated with serum resistance, tight adhesion, and production of fimbriae, respectively, functions important in micro-colony formation and potentially associated with virulence (Technical Appendix 1[PDF - 641 KB - 4 pages] Figure 1; Technical Appendix 2[PDF - 641 KB - 4 pages] Table 2) (13). The other regions of likely recombination were related to integrated prophage elements, implying H. ducreyi has an actively exchanging bacteriophage repertoire in its genome (Technical Appendix 1[PDF - 641 KB - 4 pages] Figure 1). These prophage elements included the region coding for the ctdABC genes, which have been associated with virulence (14). The class I prophage elements were absent from class II genomes but intermittently present in class I genomes (Figure 2). The presence or absence of the ctdABC coding region was not associated with cutaneous or genital ulcer disease. Another recombination region spanned the hhdA specific qPCR primer binding site. Samples with high sequence variation in this region tested negative for H. ducreyi by qPCR but gave high numbers of reads by 16S rRNA gene sequencing.
We obtained whole-genome sequences of H. ducreyi without prior culture. Most earlier studies have suggested that cutaneous strains emerged by diversification from within the class I clade (7,8), although 1 study found, in keeping with our findings, cutaneous strains emerging from class II (9). We found genital and cutaneous strains are represented in all lineages of the expanded phylogenetic tree (7). We found considerable genetic variation between class I and class II H. ducreyi sequences. Together with existing 16S rRNA data and multilocus sequence typing data (12) these findings suggest class II strains might represent a discrete subspecies of H. ducreyi.
We identified 2 samples that had been negative in the original studies but were found to contain H. ducreyi DNA in the current study. Repeated freeze–thaw cycles and limited residual DNA volumes might have contributed to our lack of detection of H. ducreyi DNA in 4 samples that tested positive in the original studies (Figure 1). Five samples that returned a weak signal by hhdAqPCR contained class II clade H. ducreyi genomes. The failure of qPCR to detect H. ducreyi in these samples was most likely explained by variation in the sequence of the hhdA pPCR primer binding sites (13) between class I and II genomes (Figure 2), demonstrating our limited understanding of the diversity of these pathogens.
Culture for H. ducreyi is not practical in the low-income settings where cutaneous strains of this organism are endemic. Next-generation sequencing circumvents this problem by enabling whole-genome sequencing direct from clinical samples. This approach considerably strengthens our ability to sequence H. ducreyi and broaden knowledge of this emerging pathogen.
Dr. Marks is an assistant professor at the London School of Hygiene & Tropical Medicine. His primary research interests are the control of neglected tropical diseases, particularly yaws and scabies.
Acknowledgment
M.M. is supported by the Wellcome Trust (102807). N.R.T. and M.F. were supported by the Wellcome Trust (098051). Funding for the fieldwork in Ghana was additionally supported by a grant from the Royal Society of Tropical Medicine & Hygiene to M.M.
References
- González-Beiras C, Marks M, Chen CY, Roberts S, Mitjà O. Epidemiology of Haemophilus ducreyi infections. Emerg Infect Dis. 2016;22:1–8. DOIPubMed
- Ghinai R, El-Duah P, Chi K-H, Pillay A, Solomon AW, Bailey RL, et al. A cross-sectional study of ‘yaws’ in districts of Ghana which have previously undertaken azithromycin mass drug administration for trachoma control. PLoS Negl Trop Dis. 2015;9:e0003496. DOIPubMed
- Mitjà O, Lukehart SA, Pokowas G, Moses P, Kapa A, Godornes C, et al. Haemophilus ducreyi as a cause of skin ulcers in children from a yaws-endemic area of Papua New Guinea: a prospective cohort study. Lancet Glob Health. 2014;2:e235–41. DOIPubMed
- Marks M, Chi K-H, Vahi V, Pillay A, Sokana O, Pavluck A, et al. Haemophilus ducreyi associated with skin ulcers among children, Solomon Islands. Emerg Infect Dis. 2014;20:1705–7. DOIPubMed
- Marks M, Mitjà O, Vestergaard LS, Pillay A, Knauf S, Chen C-Y, et al. Challenges and key research questions for yaws eradication. Lancet Infect Dis. 2015;15:1220–5. DOIPubMed
- Chen C-Y, Ballard RC. The molecular diagnosis of sexually transmitted genital ulcer disease. In: MacKenzie CR, Henrich, editors. Diagnosis of sexually transmitted diseases—methods and protocols. New York: Springer-Verlag; 2012. p. 103–12.
- Gangaiah D, Webb KM, Humphreys TL, Fortney KR, Toh E, Tai A, et al. Haemophilus ducreyi cutaneous ulcer strains are nearly identical to Class I genital ylcer strains. PLoS Negl Trop Dis. 2015;9:e0003918. DOIPubMed
- Gangaiah D, Marinov GK, Roberts SA, Robson J, Spinola SM. Draft whole-genome sequence of Haemophilus ducreyi strain AUSPNG1, isolated from a cutaneous ulcer of a child from Papua New Guinea. Genome Announc. 2016;4:e01661–15. DOIPubMed
- Gangaiah D, Spinola SM. Haemophilus ducreyi cutaneous ulcer strains diverged from both Class I and Class II genital ulcer strains: implications for epidemiological studies. PLoS Negl Trop Dis. 2016;10:e0005259. DOIPubMed
- Marks M, Vahi V, Sokana O, Puiahi E, Pavluck A, Zhang Z, et al. Mapping the epidemiology of yaws in the Solomon Islands: a cluster randomized survey. Am J Trop Med Hyg. 2015;92:129–33. DOIPubMed
- Orle KA, Gates CA, Martin DH, Body BA, Weiss JB. Simultaneous PCR detection of Haemophilus ducreyi, Treponema pallidum, and herpes simplex virus types 1 and 2 from genital ulcers. J Clin Microbiol. 1996;34:49–54.PubMed
- Quail MA, Kozarewa I, Smith F, Scally A, Stephens PJ, Durbin R, et al. A large genome center’s improvements to the Illumina sequencing system. Nat Methods. 2008;5:1005–10. DOIPubMed
- Ricotta EE, Wang N, Cutler R, Lawrence JG, Humphreys TL. Rapid divergence of two classes of Haemophilus ducreyi. J Bacteriol. 2011;193:2941–7. DOIPubMed
- Janowicz DM, Cooney SA, Walsh J, Baker B, Katz BP, Fortney KR, et al. Expression of the Flp proteins by Haemophilus ducreyi is necessary for virulence in human volunteers. BMC Microbiol. 2011;11:208. DOIPubMed
Figures
Technical Appendices
Cite This Article1These authors contributed equally to this article.






















.png)










No hay comentarios:
Publicar un comentario