
Volume 25, Number 3—March 2019
Dispatch
Whole-Genome Sequencing of Drug-Resistant Mycobacterium tuberculosis Strains, Tunisia, 2012–2016
On This Page
Imen Bouzouita , Andrea Maurizio Cabibbe, Alberto Trovato, Henda Daroui, Asma Ghariani, Basma Midouni, Leila Essalah, Emna Mehiri, Daniela Maria Cirillo, and Leila Slim Saidi
, Andrea Maurizio Cabibbe, Alberto Trovato, Henda Daroui, Asma Ghariani, Basma Midouni, Leila Essalah, Emna Mehiri, Daniela Maria Cirillo, and Leila Slim Saidi
Abstract
To investigate transmission of drug-resistant strains of Mycobacterium tuberculosis in Tunisia, we performed whole-genome sequencing on 46 multidrug-resistant strains isolated during 2012–2016. Core-genome multilocus sequence typing grouped 30 strains (65.2%) into 3 clusters, indicating extensive recent transmission and Haarlem clone predominance. Whole-genome sequencing might help public health services undertake appropriate control actions.
The emergence of drug-resistant strains of Mycobacterium tuberculosis is hampering the control of tuberculosis (TB) worldwide. In Tunisia in 2017, the estimated percentage of TB patients with multidrug-resistant (MDR)/rifampin-resistant TB was 1.1% among those with new infection and 13% among those with previously treated infection (1).
Effective and rapid tools are needed to characterize and track the transmission chains of MDR/rifampin-resistant TB. Whole-genome sequencing (WGS) has shown higher discriminatory power for epidemiologic investigations than have other conventional genotyping methods (e.g., spoligotyping, IS6110 restriction fragment length polymorphism, and mycobacterial interspersed repetitive unit–variable-number tandem repeat). Indeed, WGS has enabled investigators to rule out false transmission events (2–7). Furthermore, WGS enables simultaneous determination of polymorphisms and insertions/deletions linked to resistance to first-line and second-line drugs (8).
In this study, we used WGS to investigate transmission of MDR and extensively drug resistant (XDR) TB strains isolated in Tunisia over a 4-year period by applying the core-genome multilocus sequence typing (cgMLST) scheme and identifying the drug-resistance marker for first-line and second-line drug resistance. This study was approved by the ethics committee of A. Mami Pneumology Hospital, Ariana, Tunisia.
We retrospectively studied 46 MDR M. tuberculosis isolates collected from 46 HIV-negative patients in Tunisia during June 2012–June 2016, which represented 57 (80.7%) cases of MDR TB. Of the 46 isolates, 6 represented all (100%) XDR TB cases recorded in the country during that period. We performed drug-susceptibility testing for resistance to first-line drugs (except pyrazinamide) by using the proportion method on Lowenstein-Jensen medium. For pyrazinamide and second-line drugs, we performed drug-susceptibility testing on a Bactec MGIT 960 system (Becton, Dickinson and Company, http://www.bd.com). WGS was performed on the MiniSeq platform (Illumina Inc., https://www.illumina.com) targeting a minimum average reads coverage of 50-fold. To analyze the mutations involved in drug resistance and related to lineage determination, we used PhyResSe and TGS TB (9,10). We performed the cgMLST scheme version 2.1, considering 2,891 core genes, by using Ridom SeqSphere+ version 5.0.0 software (Ridom GmbH,https://www.cgmlst.org) (7). To define a strain as a part of a recent transmission chain, we fixed a threshold of <6 allele variants. For statistical analyses, we calculated p values by using OpenEpi version 3 (https://www.openepi.com) and considered p<0.05 to be significant.
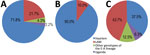
Figure 1. Lineage and family distribution of drug-resistant strains identified during study of drug-resistant Mycobacterium tuberculosis, Tunisia, 2012–2016. A) MDR strains. Haarlem was the most frequently represented family (71.8%) among MDR strains. Among...
Patient origins are reported in the Appendix. Most MDR TB cases (19; 41.3%) were recorded in the Bizerte region in northern Tunisia. WGS revealed that all MDR/XDR strains belonged to the European-American lineage (lineage 4); the Haarlem family was the most frequent (71.8%) (Figure 1). (WGS files from this study have been submitted to the European Nucleotide Archive as fastq files under study accession no. PRJEB30463, https://www.ebi.ac.uk/ena/data/search?query=PRJEB30463).
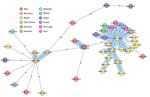
Figure 2. Core-genome multilocus sequence typing–based minimum spanning tree for strains identified during study of drug-resistant Mycobacterium tuberculosis, Tunisia, 2012–2016. Ridom SeqSphere+ minimum spanning tree for 46 samples based on 2,891 columns, pairwise...
The cluster analysis (Figure 2) showed that 30 (65.2%) of 46 isolates were grouped within 3 clusters, and most (90.0%) clustered MDR TB cases belonged to the Haarlem family (Figure 1). Of note, a big cluster of 24 MDR strains linked to Haarlem (cluster 1) was detected over the entire study period. We found no significant associations between this cluster and patient sex, age (<35 years, >35 years), and resistance to second-line drugs (XDR TB) (p>0.05); however, we found a significant association with this cluster and pyrazinamide resistance (p = 0.001) and with Bizerte (p = 0.014). Despite the association with the Bizerte region, patients from various regions were part of this cluster (Figure 2). We also found a significant association between cluster 2 (Haarlem) and Beja (p<0.001) and between cluster 3 (Latin-American-Mediterranean) and Ben Arous (p<0.001). Epidemiologic links were confirmed for patients in these 2 transmission chains (Table 1).
All cluster 1 strains shared mutations in drug-resistance genes (e.g., rpoB, Ser450Leu, the compensatory mutation rpoBVal534Met (11), katG Ser315Thr, embB Met306Ile, and gidBArg47Trp). However, diversity was noticed in pncA mutations conferring resistance to pyrazinamide. On the basis of pncAvariation, this cluster was split into 6 groups (Table 1).
Four XDR isolates belonged to cluster 1. Three XDR isolates from 2 brothers (patients 10942 and 14666) and their neighbor (patient 8626) shared mutations conferring resistance to second-line drugs and to pyrazinamide (Tables 1, 2), supporting evidence of direct transmission of this XDR strain. The fourth XDR TB case (patient/strain 13792) differed in mutations conferring resistance to pyrazinamide and fluoroquinolones. This case probably shared an ancestral MDR strain with cluster 1, which had evolved differently because of poor adherence of the patient to drug therapy (Table 2; Appendix). Five nonclustered MDR isolates linked to Haarlem (4 patients from Bizerte) were distant by 6–11 alleles from cluster 1 showing the same polymorphisms in rpoB, katG, embB, and gidB (Figure 2; Appendix). Of note, cluster 2 presented the compensatory mutation rpoB Val534Met, but no variation in embB, gidB, and pncA was detected (Appendix). This Haarlem cluster showed limited transmission compared with cluster 1, which was distant by 97 alleles (Figure 2).
In our study, mutation rpoBSer450Leu was mostly associated with rifampin resistance (n = 42, 91.3%), whereas codon 315 of katG was most involved with isoniazid resistance (n = 43, 93.4%). Three nonclustered MDR isolates presented a genomic deletion of ≈10.4 kb, which included the entire katG gene (strain 3483) and 2 uncommon mutations in katG: Gly269Asp and Gly279Asp (patients/strains 9833, 10982) (Appendix). All pyrazinamide-resistant strains detected with the MGIT 960 system (n = 33, 71.7%) had a mutation in the pncA gene or its promoter.
Regarding second-line drugs, all 13 fluoroquinolone-resistant strains had a mutation in the quinolone resistance–determining region of gyrA, gyrB, or both. For second-line injectable drugs, 4 XDR strains had the mutation rrs A1401G and 2 had the mutation eis C-14T and a frame shift in tlyA (Table 2).
cgMLST analysis showed that 65.2% of MDR/XDR strains of M. tuberculosis were clustered, reflecting extensive transmission in Tunisia, particularly of a Haarlem clone. This Haarlem clone showed polymorphisms rpoB Ser450Leu, Val534Met, katG Ser315Thr, embB Met306Ile, and gidBArg47Trp, and in pncA genes previously identified in a Haarlem MDR TB outbreak in the Bizerte region during 2001–2011 (13,14). As indicated by statistical association, we conclude that this cluster is still spreading in the Bizerte area. However, diffusion in different regions of the country is alarming and requires intensified efforts to control and diagnose drug-resistant TB.
Only 2 strains, belonging to cluster 1, did not have any mutations in pncA. The wild-type pncA isolates might represent the genotype of the first strains that emerged in Bizerte and evolved since 2001 by acquiring single-nucleotide polymorphisms in pncA and the other genes, including genes involved in resistance to second-line drugs. It has been reported that the mutation rate during a transmission chain or TB latency is not completely stable and is estimated at 0.3–0.5 single-nucleotide polymorphisms/genome/year (4,5,15), which leads to increased numbers of allele variants for some isolates and might explain the results found for 5 MDR strains distant from cluster 1 by 6–11 alleles.
The main limitation of this study is the incomplete number of MDR TB cases (≈19% missing). Epidemiologic information to confirm all clustered cases is lacking.
In summary, cgMLST-based WGS showed extensive transmission of MDR/XDR TB in Tunisia over 4 recent years, thereby indicating that MDR TB is not fully controlled. Use of this molecular approach for surveillance purposes might enable the public health service to undertake appropriate control actions, particularly in specific settings of this country.
Ms. Bouzouita is a PhD student at the National Reference Laboratory for Mycobacteria, Ariana, Tunisia. Her research focuses on TB diagnosis, TB drug resistance, and molecular epidemiology of TB.
Acknowledgment
We thank Ibtissem Blanco, Rachid Fourati, and Dhikrayet Gamara for their contribution to this study.
References
- World Health Organization. Global Tuberculosis Report. Geneva. Organization. 2018;•••:256.
Figures
Tables
Cite This ArticleOriginal Publication Date: 2/6/2019






















.png)










No hay comentarios:
Publicar un comentario