
Another Milestone in the Cystic Fibrosis Journey

Caption: Two-year-old Avalyn is among the cystic fibrosis patients who may be helped by targeted drugs.
Credit: Brittany Mahoney
Credit: Brittany Mahoney
As NIH Director, I often hear stories of how people with serious diseases—from arthritis to Zika infection—are benefitting from the transformational power of NIH’s investments in basic science. Today, I’d like to share one such advance that I find particularly exciting: news that a combination of three molecularly targeted drugs may finally make it possible to treat the vast majority of patients with cystic fibrosis (CF), our nation’s most common genetic disease.
First, a bit of history! The first genetic mutation that causes CF was discovered by a collaborative effort between my own research lab at the University of Michigan, Ann Arbor, and colleagues at the Hospital for Sick Children in Toronto—more than 25 years ago [1]. Years of hard work, supported by the National Institutes of Health and the Cystic Fibrosis Foundation, painstakingly worked out the normal function of the protein that is altered in CF, called the cystic fibrosis transmembrane regulator (CFTR). Very recently new technologies, such as cryo-EM, have given researchers the ability to map the exact structure of the protein involved in CF.
Among the tens of thousands of CF patients who stand to benefit from the next generation of targeted drugs is little Avalyn Mahoney of Cardiff by the Sea, CA. Just a few decades ago, a kid like Avalyn—who just turned 2 last month—probably wouldn’t have made it beyond her teens. But today the outlook is far brighter for her and so many others, thanks to recent advances that build upon NIH-supported basic research.
Helping to fuel this optimism are preliminary results released this week from early-stage clinical trials of three next-generation, triple combination therapies that modulate the function of CFTR. CFTR is a protein channel, indicated in gray “mesh” in the cryo-EM image below right, that regulates salt and water balance in the lungs and other parts of the body.
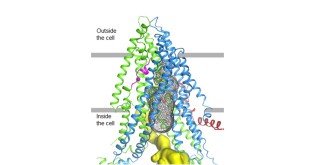
Credit: Zhang & Chen, 2016, Cell 167, 1586–1597.
People with CF have a misspelling of both copies of the CFTR gene, one inherited from each parent. There are more than 1,700 misspellings, or mutations, in the CFTR gene that can cause CF. The most common mutation, called F508del, results in a form of the CFTR protein that is misfolded and degrades prematurely. As a result, the protein fails to reach its proper location in the cell membrane, resulting in an accumulation of thick, sticky mucus that can lead to life-threatening lung infections and respiratory failure.
Developing drugs that could rescue the function of mutant CFTR has been a long and difficult journey. Initially many of us thought that gene therapy would be the way to treat this disease, but the challenges of achieving long lasting correction of the CF defect in the airways by gene transfer turned out to be extremely daunting. Beginning about 20 years ago, building on increasingly deep understanding of CFTR function derived by NIH-funded academic researchers, a partnership of the Cystic Fibrosis Foundation and a small company called Aurora (that later became Vertex) began searching for small molecules that would allow the abnormal CFTR protein to fold correctly (these are called “correctors”) and then function properly when it reached the cell membrane (these are called “potentiators”).
Now this strategy is really paying off for the 90 percent of patients who have at least one copy of F508del. According to preliminary data from one Phase 1 and two Phase 2 clinical trials reported by Vertex Pharmaceuticals Inc., Boston, on Tuesday, all three of the company’s next-gen drug combinations produced striking improvements in lung function among CF patients who have one F508del mutation and one minimal function mutation, a form of the disease that has previously been very difficult to treat. Specifically, after 2 to 4 weeks of treatment with this combination of two correctors and a potentiator, such patients experienced about a 10 percentage point improvement in forced expiratory volume in 1 second (FEV1), which is a key measure of lung capacity. Though the study was done in a double blind fashion, many participants who had been randomized to receive the active drugs quickly figured out that something very different and wonderful was happening.
Furthermore, the next-gen, combo therapies were generally well tolerated and served to improve sweat chloride levels. Elevations in sweat chloride have been used as a diagnostic test for CF for decades, and showing reductions is a powerful indicator that the drugs are working throughout the body. These results, along with data from another clinical trial expected to begin later this year, will be used by Vertex to select which next-generation targeted drug or combination of drugs will move forward into larger scale Phase III studies.
About 30,000 Americans have CF. The first major advance in molecularly targeted drug treatment for the disease came in 2012, when the Food and Drug Administration (FDA) approved ivacaftor (Kalydeco). As exciting as that was, we knew it represented just the first step on a challenging road to helping all people with CF. That’s because Kalydeco only works in the roughly 4 percent of CF patients who have the G551D mutation, along with others who have one of 23 relatively rare mutations that result in partially functioning CFTR.
The next major milestone on the therapeutic pathway was the FDA’s approval in 2015 of Orkambi, a drug that combines ivacaftor with lumacaftor, the first “corrector”. That combination therapy was designed to help CF patients with two copies of the F508del mutation. Another combination therapy (ivacaftor plus tezacaftor) aimed at CF caused by two copies of the F508del mutation is currently under review by the FDA.
All told, there is now some sort of approved targeted treatment for about 40 percent of people with CF. However, if the new triple-drug combos live up to their early promise, Michael Boyle, senior vice president of the Cystic Fibrosis Foundation, Bethesda, MD, thinks it may be possible to treat up to 90 percent of people with CF. That’s fantastic news, but I can tell you that neither the Cystic Fibrosis Foundation nor the NIH will rest until there are effective and affordable treatments—or, better yet, a cure—for each and every person with CF, both here in the United States and around the globe.
Reference:
[1] Identification of the cystic fibrosis gene: chromosome walking and jumping. Rommens JM1, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N, et al. Science. 1989 Sep 8;245(4922):1059-1065.
Links:
Positive Early Study Results for Next-Generation CFTR Modulators. Cystic Fibrosis Foundation News Release, July 18, 2017.
What is Cystic Fibrosis (National Heart, Lung, and Blood Institute/NIH)
Genetics Home Reference: Cystic Fibrosis (National Library of Medicine/NIH)




















.png)












No hay comentarios:
Publicar un comentario