
Volume 26, Number 7—July 2020
Research
Efficient Surveillance of Plasmodium knowlesi Genetic Subpopulations, Malaysian Borneo, 2000–2018
Article Metrics
Paul C.S Divis1 , Ting H. Hu1, Khamisah A. Kadir, Dayang S.A. Mohammad, King C. Hii, Cyrus Daneshvar, David J. Conway, and Balbir Singh
, Ting H. Hu1, Khamisah A. Kadir, Dayang S.A. Mohammad, King C. Hii, Cyrus Daneshvar, David J. Conway, and Balbir Singh
Abstract
Population genetic analysis revealed that Plasmodium knowlesi infections in Malaysian Borneo are caused by 2 divergent parasites associated with long-tailed (cluster 1) and pig-tailed (cluster 2) macaques. Because the transmission ecology is likely to differ for each macaque species, we developed a simple genotyping PCR to efficiently distinguish between and survey the 2 parasite subpopulations. This assay confirmed differences in the relative proportions in areas of Kapit division of Sarawak state, consistent with multilocus microsatellite analyses. Analyses of 1,204 human infections at Kapit Hospital showed that cluster 1 caused approximately two thirds of cases with no significant temporal changes from 2000 to 2018. We observed an apparent increase in overall numbers in the most recent 2 years studied, driven mainly by increased cluster 1 parasite infections. Continued monitoring of the frequency of different parasite subpopulations and correlation with environmental alterations are necessary to determine whether the epidemiology will change substantially.
The monkey parasite Plasmodium knowlesi was discovered to be a common cause of malaria in humans in 2004, initially from investigations in the Kapit division of Sarawak state, Malaysian Borneo (1). Humans acquire infection primarily from wild long-tailed (Macaca fascicularis) and pig-tailed (M. nemestrina) macaque reservoirs (2); Anopheles mosquitoes of the leucosphyrus group are vectors (3,4). P. knowlesi malaria has been described across Southeast Asia, but most clinical cases are still reported in Malaysian Borneo (3,5–8). In 2017 and 2018, a total of 7,745 cases were reported in Malaysia, 86.8% of which were detected in Malaysian Borneo (B. Singh, unpub. data) (9). P. knowlesi infections can be asymptomatic (10,11), and clinical cases exhibit a wide spectrum of disease ranging from mild symptoms to death (3).
Population genetic surveys of P. knowlesi infections in humans across Malaysia have revealed 2 divergent subpopulations of the parasite in Malaysian Borneo that are associated with the 2 macaque species locally, suggesting 2 independent zoonoses (12,13). The cluster 1 type has been associated with long-tailed macaques and the cluster 2 type with pig-tailed macaques (12). The existence of 2 sympatric subpopulations also has been confirmed by whole-genome sequencing (WGS) of P. knowlesi from patients in Malaysian Borneo (13,14). In peninsular Malaysia on the Asia mainland, all cases have been caused by another subpopulation, cluster 3, that has not been detected in Malaysian Borneo (13,15). Limited WGS (14,15) and microsatellite (13) genotyping of P. knowlesi isolates derived from human and only long-tailed macaque hosts from peninsular Malaysia showed allopatric divergence for this subpopulation cluster from those of Malaysian Borneo because of geographic separation by the South China Sea.
Increasing numbers of P. knowlesi malaria cases detected might be due to increased zoonotic exposure along with a reduction of endemic malaria parasite species (16). With the recent identification of different zoonotic P. knowlesi genetic subpopulations, determining whether these populations vary in frequency over space and time is important. Interactions with vectors and reservoir hosts will affect distributions of these parasite subpopulations, and human contacts with these vectors and reservoir hosts would determine zoonotic incidence. Because the previously used microsatellite genotyping and WGS are time consuming and expensive, we developed a simple PCR to discriminate between the 2 P. knowlesi subpopulations in Malaysian Borneo.
Identification of Cluster-Specific Markers
We identified high-quality bi-allelic single-nucleotide polymorphisms (SNPs) from whole-genome sequence data of P. knowlesi isolates from cluster 1 (n = 38) and cluster 2 (n = 10) subpopulations (15). We identified 9,293 SNPs with complete fixation of alternative alleles between the 2 subpopulations. Oligonucleotides for allele-specific PCRs were designed on the basis of genome regions with a high density of fixed SNPs and having multiple SNPs <5 nt apart, especially at the 3′ ends of the designed primers. For each subpopulation, we chose 5 pairs of forward and reverse PCR primers to evaluate for genotyping assays (Appendix Table 1).
Cluster-Specific Genotyping PCR
PCRs to discriminate subpopulation clusters were optimized in 11-μL reaction volume containing 1X Green GoTaq Flexi buffer (Promega, ), 5 mmol/L MgCl2, 0.275 U GoTaq DNA polymerase, 0.2 mmol/L dNTP (Bioline, ), 1–2-μL DNA template, and 0.25 μmol/L each forward and reverse primers. We tested 2 thermal cycling methods to determine the optimum annealing temperature. The first conventional method was 94°C for 2 min, followed by 35 cycles of initial denaturation of 94°C for 30 s, gradient annealing from 50°C to 66°C for 1 min, extension at 72°C for 30 s, and final elongation at 72°C for 2 min. For the second method, we used the touchdown PCR to increase the specificity and sensitivity in amplification (17) with the following 2 phases. We initiated the first phase with denaturation at 94°C for 2 min, followed by 10 cycles of touchdown program of 94°C for 30 s, annealing of 10°C above the primer melting temperature for 45 s and 72°C for 30 s, with a decreased on 1°C of the annealing temperature every cycle. Then, we performed an additional 25 cycles as follows: 94°C for 30 s, annealing of 5°C below the primer annealing primer melting temperature for 45 s, 72°C for 30 s, and final elongation at 72°C for 5 min. We analyzed PCR products by electrophoresis on agarose gel stained with ethidium bromide and visualized them under ultraviolet transillumination.
DNA and Blood Samples from Malaria Infections
To validate allele-specific PCRs, we obtained DNA samples of P. knowlesi infections in Malaysian Borneo from previous studies (12,13,15). Samples from humans were from Betong (n = 29), Kanowit (n = 34), Miri (n = 46), Sarikei (n = 23), Kapit (n = 52), Kudat (n = 46), Ranau (n = 62), and Tenom (n = 48); we also used samples from long-tailed macaques (n = 10) and pig-tailed macaques (n = 5) from Kapit (2). We determined subpopulation assignments (cluster 1 or cluster 2) of each infection by analyzing multilocus microsatellite data (12) or whole-genome sequence data (15). Plasmodium DNA controls of humans (P. falciparum, P. vivax, P. malariae, and P. ovale) and macaques (P. knowlesi, P. inui, P. cynomolgi, P. fieldi, and P. coatneyi) were also used for specificity tests of the PCRs.
To determine the frequency of the 2 P. knowlesi subpopulations over time, we investigated samples derived from Kapit division, Sarawak state, Malaysian Borneo, where high incidence of cases has been reported (7). New samples were collected as dried blood spots on filter papers (3MM Whatman, ) from P. knowlesi clinical cases in Kapit Hospital during September 2016–May 2018 (440 cases). DNA was extracted and P. knowlesi confirmed by species-specific nested PCRs (2) at the Malaria Research Centre, Universiti Malaysia Sarawak (UNIMAS, Kota Samarahan, Malaysia). We also analyzed 764 DNA samples from persons with P. knowlesi infection at Kapit Hospital collected previously: March 2000–December 2002, 110 samples (1); March 2006–February 2008, 176 samples (18); and June 2013–mid-September 2016, 478 samples (13).
Data Analysis and Ethics Approval
We tabulated genotyping data in Excel and conducted statistical analysis using R (). We also explored temporal patterns for overall P. knowlesi subpopulation cases over the previous 5 years using the R package seasonal decomposition of time series (STL) by LOESS (locally estimated scatterplot smoothing) (19). We generated STL decomposition using periodic as smoothing parameter for the seasonal component (ns) with no robustness iterations. LOESS is the seasonal-trend decomposition method used in estimating trend from the time series data. The Medical Ethics Committees of UNIMAS (NC-21.02/03-02 Jld 2 [19]) and the Medical Research and Ethics Committee, Malaysian Ministry of Health (NMRR-16-943-31224), approved this study.
Development of Simple PCR to Discriminate P. knowlesi Cluster 1 and 2 Subpopulations

Figure 1. Specificity of PCR primer sets C1A and C2J for discriminating Plasmodium knowlesi infections of cluster 1 and cluster 2 subpopulations, Kapit division, Sarawak state, Malaysian Borneo. Lane 1, DNA ladder; lane...
We tested 10 PCRs on the basis of fixed SNP differences for performance in discriminating the 2 sympatric P. knowlesi subpopulations. Of these, 1 assay each for cluster 1 (primer pair C1A) and cluster 2 (primer pair C2J) showed complete specificity when tested on 5 cluster 1 and 9 cluster 2 DNA samples (Figure 1; Appendix Table 2) that were previously confirmed by microsatellites cluster assignments (13). These primers were also P. knowlesi–specific and did not show amplification on DNA of other simian malaria parasites (P. coatneyi, P. cynomolgi, P. fieldi, and P. inui) or human malaria parasites (P. falciparum, P. vivax, P. malariae, and P. ovale), or host DNA from humans or either macaque host species (Appendix Figure 1).
We further tested these primer sets for sensitivity using pure DNA of cluster 1 samples with starting parasitemias of 13,793 parasites/μL blood and cluster 2 samples with starting parasitemias of 8,017 parasites/μL blood. We prepared 5-fold serial dilutions to serve as template in separate PCRs. Tests of sensitivity showed limit of detection at ≈4 parasites/μL blood for primer pair C1A and ≈13 parasites/μL blood for primer pair C2J (Appendix Figure 2).
Proportions of P. knowlesi Subpopulations in Different Areas
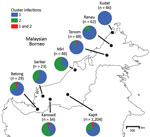
Figure 2. Proportions of cluster 1 and cluster 2 Plasmodium knowlesi infections as defined with the cluster-specific PCRs, Malaysian Borneo. A total of 1,492 P. knowlesi infections from humans were genotyped using the...
Analysis of DNA samples of 1,492 P. knowlesi malaria cases in different regions of Malaysian Borneo showed cluster 1 as the predominant subpopulation (70%), followed by cluster 2 (28%) (Figure 2; Appendix Table 3). Only 2% of infections were positive for both primer sets, and these infections had mixed genotypes as identified by previous microsatellite analysis (12,13). Cluster 1 infections were more common at most individual locations sampled throughout Malaysian Borneo, except for Miri and Kanowit, where cluster 2 infections were more common.
We also tested the primer sets on 15 natural infections of wild macaques (10 long-tailed and 5 pig-tailed) from Kapit. P. knowlesi infections from all macaques showed concordant macaque host association (cluster 1 in long-tailed macaques and cluster 2 in pig-tailed macaques), except for 1 long-tailed macaque that was infected by both cluster 1 and cluster 2 parasites (Appendix Table 3).
Temporal Analysis of P. knowlesi Subpopulation Types in a High-Incidence Area

Figure 3. Proportions of Plasmodium knowlesi subpopulations in humans of the Kapit division, Sarawak state, Malaysian Borneo, 2000–2018. A total of 1,204 P. knowlesi infections were genotyped, and distribution patterns of subpopulation clusters...
In Kapit, where a high incidence of cases has been reported since the zoonosis was discovered to be common (1,7,18), we analyzed DNA samples from 1,204 human P. knowlesi infections collected in 3 study periods during 2000–2018. Of those infections, 69% belonged to the cluster 1 subpopulation (Figure 3, panel A; Appendix Table 4). Most (28%) of the remaining P. knowlesi infections belonged to the cluster 2 subpopulation, and only 2% had parasites of both types. During 3 study periods of 2000–2002 (n = 110 cases), 2006–2008 (n = 176 cases), and 2013–2018 (n = 918 cases), the proportion of cluster 1 and cluster 2 subpopulations showed similar patterns of distribution with cluster 1 subpopulation as the predominant ones (Pearson p = 0.74). We also observed similar patterns across 12 months of these study periods (Figure 3, panel B), indicating a lack of strong seasonal variation between cluster 1 and cluster 2 infections in Kapit (p = 0.56 by Fisher exact test).
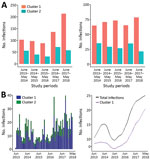
Figure 4. Frequency and trend line patterns of Plasmodium knowlesi subpopulations in Kapit division, Sarawak state, Malaysian Borneo, June 2013–May 2018. A) Distribution of 637 cluster 1 and 258 cluster 2 infections during...
We conducted a more intensive analysis on infections sampled during the most recent 5-year study period (June 2013–May 2018); we made a particular effort to recruit most of the case-patients seeking care at Kapit Hospital during this time. Of the 918 infections genotyped from this period, 637 were cluster 1 infections, 258 were cluster 2 infections, and 23 were mixed cluster infections. The proportion of cluster 1 infections always ranged from 63% to 78% (Pearson p = 0.007) and was highest in the most recent year. The total number of P. knowlesi cases was also highest in the most recent year (Figure 4, panel A). Using an STL decomposition method of analysis based on numbers plotted on a monthly basis (Figure 4, panel B), we noted a trend of increasing numbers of cases overall, as well as of the cluster 1 subpopulation separately, from June 2016 onward. We found no significant trend for cluster 2 cases separately.
On the basis of fixed SNP differences between the 2 P. knowlesi genetic subpopulations in Malaysian Borneo, identified by whole-genome sequence data analysis (14,15), we successfully developed an allele-specific PCR for discriminating these in large-scale studies. This method enables identification of cluster 1 and cluster 2 subpopulations from field isolates throughout the sympatric distribution, without the need to perform population genetic analysis of multilocus data on all samples. A different P. knowlesi subpopulation termed cluster 3 has been described to account for all cases in Peninsular Malaysia (13,15), but we did not study that subpopulation because it has not been detected in Malaysian Borneo.
The use of these allele-specific PCRs is limited to P. knowlesi infections in Malaysian Borneo because they were designed on the basis of the genome sequences of the cluster 1 and cluster 2 parasites found there. These assays could potentially be applied to analyze P. knowlesi infections in Kalimantan, Indonesian Borneo, which shares international borders with Malaysian Borneo. The allopatric divergence of cluster 3 subpopulations in Peninsular Malaysia (13) indicates the need for development of a separate allele-specific PCR for those parasites, which would require analysis of whole-genome data from substantially more P. knowlesi isolates from Peninsular Malaysia.
The validation of this allele-specific PCR shows remarkable sensitivity with single-round (nonnested) amplification, even at a parasitemia of ≈4 parasites/μL blood. Although all samples tested from patients at Kapit Hospital could be genotyped, this PCR would have limitations on subpatent P. knowlesi infections when parasites are undetectable under the microscope (10,11,20).
No clear evidence of direct human–mosquito–human transmission of P. knowlesi has yet been detremined, although such transmission might be occurring. Most hospitalized P. knowlesi malaria patients have been adult farmers or logging camp workers, who regularly spend time in forests or forest fringes, or travelers entering forests (3), suggesting that macaque–vector–human transmission is the primary route of infection (21). Although the distribution of long-tailed and pig-tailed macaques overlaps throughout Southeast Asia, these macaque species show different habitat preferences (22). The preference of long-tailed macaques for cropland, wetland, and urban areas brings them in close proximity to humans, but whether transmission occurs outside of the forests is not known. The widespread distribution of long-tailed macaques corresponds with the cluster 1 parasite subpopulation accounting for most P. knowlesi infections in Kapit. The lower frequency of the cluster 2 subpopulation in humans is as expected, given that those parasites are associated with pig-tailed macaques, which occur in more remote forested areas (22).
Like many other vectorborne parasitic diseases, malaria is sensitive to environmental changes such as deforestation (23); the association between incidences of P. knowlesi infections and environmental changes in Sabah, Malaysian Borneo, supports this fact (16,24), because overall numbers of cases have increased in the past few years (8). We have shown that most of the cases in Sabah are of the cluster 1 genetic subpopulation of P. knowlesi, and that numbers of this parasite subpopulation have increased in Kapit in the most recent 2 years of the study period. Whether changes in the landscape in Sarawak or other factors have contributed to the increasing numbers of P. knowlesi cases needs to be studied. Determining whether any significant differences exist between the clinical outcomes after infection with the different subpopulations also is important. Such determination requires collecting detailed clinical and laboratory data on P. knowlesi patients infected with the 2 different subpopulation clusters and performing careful association study analysis to deliver robust inference for the benefits of both clinicians and public health experts.
P. knowlesi infections require separate efforts from those being targeting elimination of human malaria (P. falciparum and P. vivax), particularly in Malaysia, where zoonotic malaria is dominant (25). Although P. knowlesi is not part of the national elimination program (26), monitoring this parasitic infection is crucial because of its increasing incidence. Zoonotic malaria requires new strategies in prevention and control, including monitoring of the different parasite genetic subpopulations.
Acknowledgments
We thank colleagues at the Malaria Research Centre of UNIMAS and the London School of Hygiene and Tropical Medicine, as well as medical laboratory technologists in Kapit Hospital, Sarawak, for laboratory assistance. We also thank the Director General of Health in Malaysia for permission to publish this article.
This study is supported by postgraduate scholarship from the Sarawak Scholarship Foundation and Ministry of Higher Education in Malaysia, and research grants from UNIMAS (grants nos. F05/SPTDG/1447/16/4, F05/DPP/1505/2016, and F05/SpGS/1551/2017).
Dr. Divis is a senior lecturer at the Faculty of Medicine and Health Sciences and a research fellow at the Malaria Research Centre in UNIMAS, Malaysia. His research interests include molecular epidemiology, population genetics, and genomics of P. knowlesi.
References
- Singh B, Kim Sung L, Matusop A, Radhakrishnan A, Shamsul SS, Cox-Singh J, et al. A large focus of naturally acquired Plasmodium knowlesi infections in human beings. Lancet. 2004;363:1017–24.
- Lee KS, Divis PC, Zakaria SK, Matusop A, Julin RA, Conway DJ, et al. Plasmodium knowlesi: reservoir hosts and tracking the emergence in humans and macaques. PLoS Pathog. 2011;7:
e1002015 . - Singh B, Daneshvar C. Human infections and detection of Plasmodium knowlesi. Clin Microbiol Rev. 2013;26:165–84.
- Vythilingam I, Lim YA, Venugopalan B, Ngui R, Leong CS, Wong ML, et al. Plasmodium knowlesi malaria an emerging public health problem in Hulu Selangor, Selangor, Malaysia (2009-2013): epidemiologic and entomologic analysis. Parasit Vectors. 2014;7:436.
- William T, Rahman HA, Jelip J, Ibrahim MY, Menon J, Grigg MJ, et al. Increasing incidence of Plasmodium knowlesi malaria following control of P. falciparum and P. vivax Malaria in Sabah, Malaysia. PLoS Negl Trop Dis. 2013;7:
e2026 . - Yusof R, Lau YL, Mahmud R, Fong MY, Jelip J, Ngian HU, et al. High proportion of knowlesi malaria in recent malaria cases in Malaysia. Malar J. 2014;13:168.
- Ooi CH, Bujang MA, Tg Abu Bakar Sidik TMI, Ngui R, Lim YA. Over two decades of Plasmodium knowlesi infections in Sarawak: Trend and forecast. Acta Trop. 2017;176:83–90.
- Cooper DJ, Rajahram GS, William T, Jelip J, Mohammad R, Benedict J, et al. Plasmodium knowlesi malaria in Sabah, Malaysia, 2015–2017: ongoing increase in incidence despite near-elimination of the human-only Plasmodium species. Clin Infect Dis. 2020;70:361–7.
- World Health Organization. World malaria report. Geneva: The Organization; 2018.
- Fornace KM, Nuin NA, Betson M, Grigg MJ, William T, Anstey NM, et al. Asymptomatic and submicroscopic carriage of Plasmodium knowlesi malaria in household and community members of clinical cases in Sabah, Malaysia. J Infect Dis. 2016;213:784–7.
- Lubis IND, Wijaya H, Lubis M, Lubis CP, Divis PCS, Beshir KB, et al. Contribution of Plasmodium knowlesi to multi-species human malaria infections in North Sumatera, Indonesia. J Infect Dis. 2017;215:1148–55.
- Divis PC, Singh B, Anderios F, Hisam S, Matusop A, Kocken CH, et al. Admixture in humans of two divergent Plasmodium knowlesi populations associated with different macaque host species. PLoS Pathog. 2015;11:
e1004888 . - Divis PC, Lin LC, Rovie-Ryan JJ, Kadir KA, Anderios F, Hisam S, et al. Three divergent subpopulations of the malaria parasite Plasmodium knowlesi. Emerg Infect Dis. 2017;23:616–24.
- Divis PCS, Duffy CW, Kadir KA, Singh B, Conway DJ. Genome-wide mosaicism in divergence between zoonotic malaria parasite subpopulations with separate sympatric transmission cycles. Mol Ecol. 2018;27:860–70.
- Assefa S, Lim C, Preston MD, Duffy CW, Nair MB, Adroub SA, et al. Population genomic structure and adaptation in the zoonotic malaria parasite Plasmodium knowlesi. Proc Natl Acad Sci U S A. 2015;112:13027–32.
- Fornace KM, Abidin TR, Alexander N, Brock P, Grigg MJ, Murphy A, et al. Association between landscape factors and spatial patterns of Plasmodium knowlesi infections in Sabah, Malaysia. Emerg Infect Dis. 2016;22:201–8.
- Korbie DJ, Mattick JS. Touchdown PCR for increased specificity and sensitivity in PCR amplification. Nat Protoc. 2008;3:1452–6.
- Daneshvar C, Davis TM, Cox-Singh J, Rafa’ee MZ, Zakaria SK, Divis PC, et al. Clinical and parasitological response to oral chloroquine and primaquine in uncomplicated human Plasmodium knowlesi infections. Malar J. 2010;9:238.
- Cleveland RB, Cleveland WS, McRae JE, Terpenning I. STL: a seasonal-trend decomposition procedure based on LOESS. J Off Stat. 1990;6:3–73.
- Siner A, Liew ST, Kadir KA, Mohamad DSA, Thomas FK, Zulkarnaen M, et al. Absence of Plasmodium inui and Plasmodium cynomolgi, but detection of Plasmodium knowlesi and Plasmodium vivax infections in asymptomatic humans in the Betong division of Sarawak, Malaysian Borneo. Malar J. 2017;16:417.
- Imai N, White MT, Ghani AC, Drakeley CJ. Transmission and control of Plasmodium knowlesi: a mathematical modelling study. PLoS Negl Trop Dis. 2014;8:
e2978 . - Moyes CL, Shearer FM, Huang Z, Wiebe A, Gibson HS, Nijman V, et al. Predicting the geographical distributions of the macaque hosts and mosquito vectors of Plasmodium knowlesi malaria in forested and non-forested areas. Parasit Vectors. 2016;9:242.
- Confalonieri UE, Margonari C, Quintão AF. Environmental change and the dynamics of parasitic diseases in the Amazon. Acta Trop. 2014;129:33–41.
- Brock PM, Fornace KM, Grigg MJ, Anstey NM, William T, Cox J, et al. Predictive analysis across spatial scales links zoonotic malaria to deforestation. Proc Biol Sci. 2019;286:
20182351 . - Barber BE, Rajahram GS, Grigg MJ, William T, Anstey NM. World Malaria Report: time to acknowledge Plasmodium knowlesi malaria. Malar J. 2017;16:135.
- World Health Organization. Eliminating malaria: case study 8. Progress towards elimination in Malaysia. San Francisco: University of California; 2015.
Figures
Cite This ArticleOriginal Publication Date: June 11, 2020
1These authors contributed equally to this article.




















.png)


.png)










No hay comentarios:
Publicar un comentario