.jpg)
A map of SARS-CoV-2 escape mutations enables the development of therapeutic antibody cocktails

A recent tour-de-force study by the US researchers describes a comprehensive approach to completely map mutations to the SARS-CoV-2 receptor-binding domain (RBD) that escape antibody binding – enabling, in turn, rational design of antibody therapeutics and appraisal of the antigenic consequences of viral evolution. The results are currently available in a bioRxiv* preprint paper.
The coronavirus disease (COVID-19) pandemic, caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has prompted urgent and increased interest in antibody treatment options and especially vaccines that induce effective antibodies against the virus.
A wide array of potent neutralizing anti-SARS-CoV-2 antibodies target RBD of the viral spike glycoprotein, frequently competing with its binding to the angiotensin-converting enzyme 2 (ACE2 receptor). Furthermore, anti-RBD antibodies often govern the neutralizing activity of the polyclonal antibody response after natural infection.
Both passively delivered and vaccine-induced anti-RBD neutralizing antibodies safeguard against SARS-CoV-2 in animal models; nonetheless, preliminary evidence implies that the presence of neutralizing antibodies can also be linked with protection in humans.
A yeast-display system to completely map SARS-CoV-2 RBD antibody escape mutations. (A) Yeast display RBD on their surface. The RBD contains a c-myc tag, enabling dual-fluorescent labeling to quantify both RBD expression and antibody binding of RBD by flow cytometry. (B) Per-cell RBD expression and antibody binding as measured by flow cytometry for yeast expressing unmutated RBD and one of the RBD mutant libraries. (C) Experimental workflow. Yeast expressing RBD mutant libraries are sorted to purge RBD mutations that abolish ACE2 binding or RBD folding. These mutant libraries are then labeled with antibody, and cells expressing RBD mutants with decreased antibody binding are enriched using FACS (the “antibody-escape” bin; see Figure S1 for gating details). Both the initial and antibody-escape populations are deep sequenced to identify mutations enriched in the antibody-escape population. The deep-sequencing counts are used to compute the “escape fraction” for each mutation, which represents the fraction of yeast cells with a given RBD mutation that falls into the antibody-escape sort bin. The escape fractions are represented in logo plots, with tall letters indicating mutations that strongly escape antibody binding.
The rise of viral mutants
Consequently, ascertaining which viral mutations escape from antibodies becomes pivotal for designing treatment options and vaccines, but also for evaluating the antigenic implications of viral evolution. Previous studies have shown that such mutations could become dominant over a longer evolutionary period.
Still, currently available methods to pinpoint SARS-CoV-2 escape mutations by passaging the viral agent in the presence of antibodies are deficient, primarily in the sense that they only find one or a handful of possible escape mutations.
A research group, led by Dr. Allison J. Greaney from the Fred Hutchinson Cancer Research Center and the University of Washington in Seattle, overcame these limitations by introducing a high-throughput approach to completely map mutations that elude antibody binding in the SARS-CoV-2 RBD and applied this approach to 10 human antibodies.
Mapping SARS-CoV-2 antibody-escape mutations
More specifically, to map antibody-escape mutations in such manner, these scientists leveraged a system for expressing conformationally-intact RBD on the exterior of yeast cells. Furthermore, they have devised duplicate mutant libraries of the RBD from the Wuhan-Hu-1 SARS-CoV-2 strain to pinpoint mutations that allow the great escape from antibodies.
Then they have applied this escape-mutation mapping to 10 human monoclonal antibodies: 9 neutralizing antibodies that were isolated from convalescent COVID-19 patients, and a recombinant form of one cross-reactive non-neutralizing antibody that was isolated from a convalescent patient with the original SARS infection (i.e., SARS-CoV-1).
In an effort to improve the comparison of the escape maps across antibodies, multidimensional scaling was used to summarize escape mutations similarities into a two-dimensional plot. Experimentally, key escape mutations in neutralization assays were tested with the use of spike-pseudotyped lentiviral particles to validate the escape maps.
Finally, the researchers have used negative-stain electron microscopy to examine the extent to which the escape maps could be rationalized regarding the three-dimensional structures of the antibody-RBD complexes.
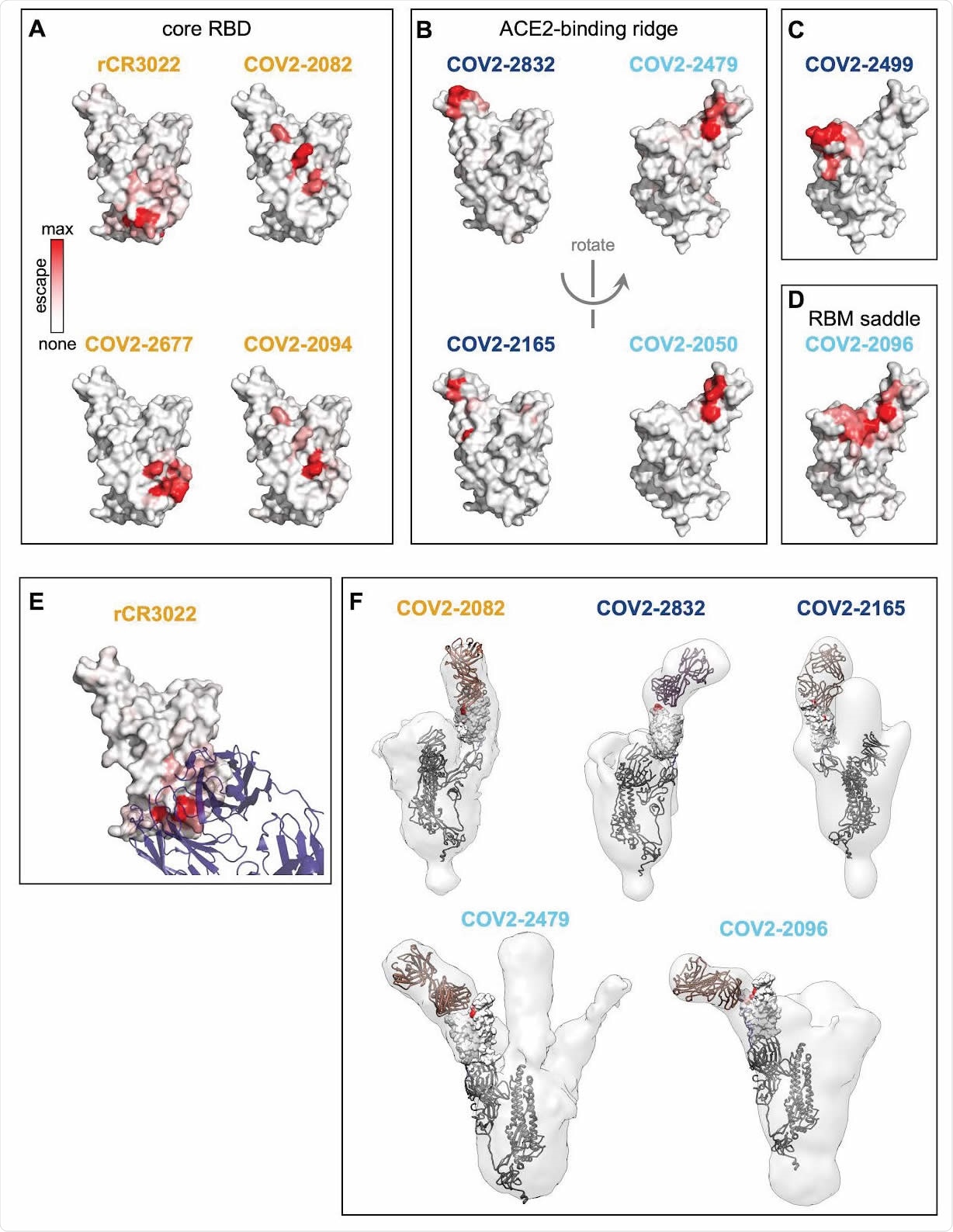
Structural mapping of antibody binding and escape. (A-D) For each antibody, the structure shows the RBD surface (PDB 6M0J) colored by the largest-effect escape mutation at each site, with white indicating no escape and red indicating the strongest escape mutation for that antibody. Antibodies are arranged so that those with similar structural epitopes are in the same panel, namely by whether their epitopes are in (A) the core of the RBD, (B) the ACE2-binding ridge, (C) the opposite edge of the RBM, or (D) the saddle of the RBM surface. (E) Crystal structure of the rCR3022-bound RBD (PDB 6W41), with Fab in purple and RBD colored according to sites of escape as in (A). (F) For 5 monoclonal antibodies, Fab bound to SARS-CoV-2 spike ectodomain trimer was visualized by negative-stain electron microscopy (EM). The RBD is modeled as a surface representation, colored according to sites of escape as in (A). Fab chains are modeled in gold. Antibody names are colored according to Figure 2B: core-binding, orange; RBM-binding, cyan; ACE2 contact site-binding, dark blue. See https://jbloomlab.github.io/SARS-CoV-2-RBD_MAP_Crowe_antibodies/ for interactive versions of the escape-colored structures in (A-D).
Nuances of high affinity binding
In this study, all ten antibodies bound the SARS-CoV-2 RBD with high affinity; however, they differed in the scope of competition with ACE2 for RBD binding, their neutralization potencies, as well as cross-reactivity with SARS-CoV-1.
These maps definitely complement thus far dominant structure-based approaches that outline the physical interface between the virus and an antibody but do not directly measure the effect of mutations on antibody binding.
There are remarkable nuances in which mutations escape individual antibodies captured by this research endeavor. Moreover, the fine details of the escape maps reveal that the effects of specific mutations can vary substantially – even among antibodies that seemingly target the same region.
The rational design of antibody cocktails
Counter to familiar wisdom, antibody cocktails do not necessarily have to target different regions of the RBD in order to withstand viral escape. A simple inspection of the escape maps demonstrates pairs of antibodies targeting the RBD's ACE2-binding interface without common escape mutations.
This means that complete escape-mutation maps basically open the door for the rational design of antibody therapeutics, as well as the assessment of the antigenic repercussions of viral evolution. In the meantime, the extent to which mutations that drastically affect the antigenicity of SARS-CoV-2 will persist during viral evolution remains an open question.
*Important Notice
bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
Journal reference:
- Greaney, A.J. et al. (2020). Complete mapping of mutations to the SARS-CoV-2 spike receptor-binding domain that escape antibody recognition. bioRxiv. https://doi.org/10.1101/2020.09.10.292078.




















.png)


.png)










No hay comentarios:
Publicar un comentario